Neue EU-Verordnungen: Die wichtigsten Neuerungen im Überblick

DI Martin Schmid, Geschäftsführer en.co.tec
Vertreter der 28 Mitgliedstaaten im EU-Ministerrat haben am 07.03.2017 den Vorschlag für die neue europäische Gesetzgebung über Medizinprodukte und In-Vitro-Diagnostika angenommen. Das EU-Parlament wird im April die beiden Verordnungen verabschieden, danach werden sie im Amtsblatt veröffentlicht. Die Verordnung tritt dann am zwanzigsten Tag nach ihrer Veröffentlichung in Kraft (also voraussichtlich im Mai / Juni 2017).

Abb.: Zeitschiene vom ersten Kommissionsvorschlag bis heute
Übergangsfristen
Die Richtlinien für Medizinprodukte (93/42/EWG) und Aktive Implantierbare Medizinprodukte (90/385/EWG) werden zu einer Medizinprodukte-Verordnung zusammengeführt und verlieren nach einer Übergangsfrist von 3 Jahren ihre Gültigkeit. Ab ca. Mai 2020 ist dann die Medizinprodukte-Verordnung allein gültig.
Die Richtlinie für In-vitro-Diagnostika (98/79/EG) verliert nach einer Übergangsfrist von 5 Jahren ihre Gültigkeit. Ab ca. Mai 2022 ist dann die In-vitro-Diagnostika-Verordnung allein gültig.
Weitere Übergangsfristen finden Sie in Artikel 120 (MDR) und Artikel 110 (IVDR).
Besonders wichtig ist, dass es bei Geltungsbeginn der Verordnungen eine ausreichende Zahl von Benannten Stellen (gemäß den neuen Bestimmungen) gibt, damit Marktengpässe wie aktuell vermieden werden.
Vergleich bisherige Richtlinien & neue MDR / IVDR
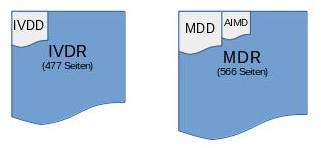
 Die Verordnungen sind im Vergleich zu den Richtlinien wesentlich umfangreicher (MDR: 566 Seiten, MDD 65 Seiten). Die MDR umfasst neben 67 Seiten Erwägungsgründen (bisher MDD 5 Seiten) 123 Artikel (bisher MDD 23) und 17 Anhänge (bisher MDD 10).
Die Verordnungen sind im Vergleich zu den Richtlinien wesentlich umfangreicher (MDR: 566 Seiten, MDD 65 Seiten). Die MDR umfasst neben 67 Seiten Erwägungsgründen (bisher MDD 5 Seiten) 123 Artikel (bisher MDD 23) und 17 Anhänge (bisher MDD 10).- Die Inhalte von MDR und IVDR sind nun viel stärker harmonisiert. So sind beispielsweise auch In-vitro-Diagnostika einer risikobasierten Klassifizierung* zu unterziehen. Das bedeutet einen Paradigma-Wechsel und wird v.a. IVD-Hersteller, deren Produkte bisher allgemeine IVD’s waren, vor fundamentale Herausforderungen stellen. (*d.h.: Qualitätsmanagement-System + Benannte Stelle).
- Erklärtes Ziel der neuen Verordnung ist die Gewährleistung der Sicherheit bei gleichzeitiger, schneller Versorgung der Patienten mit innovativen Medizinprodukten. Auch wenn einige äußerst kritische Punkte aus der neuen Medizinprodukteverordnung verbannt werden konnten, enthält die Endfassung dennoch zahlreiche kritische Herausforderungen für die Hersteller: Unter anderem die Höherklassifizierungen von Produkten (z.B.: Regel 11 zur Klassifizierung von Software, oder stoffliche Medizinprodukte) sowie der verschärfte Marktzugangsprozess (Scrutiny) für neue, implantierbare Produkte der Klasse III und Klasse IIb-Produkte, welche Arzneimittel zuführen.
Viele neue Herausforderungen für Medizinproduktehersteller
Die neue EU-Verordnung verschärft die Vorschriften, um Medizinprodukte auf den Markt zu bringen, und verstärkt die Markt-Überwachung von Medizinprodukten.
Die Verordnung wird auch die Vorschriften für benannte Stellen und die Überwachung dieser verschärfen. Benannte Stellen werden dazu angehalten, unangekündigte Audits in den Unternehmen durchzuführen.
Die neue Verordnung enthält explizite Bestimmungen zur Verantwortung des Herstellers für die Rückverfolgbarkeit von Qualität, Leistung und Sicherheit der Medizinprodukte, die bereits am Markt sind. Dies ermöglicht es Herstellern, rasch zu handeln, wenn Bedenken bestehen und soll Herstellern auch helfen, auf der Grundlage dieser Daten ihre Produkte kontinuierlich zu verbessern.
Hersteller werden klare Verantwortlichkeiten, z.B. für Haftung, aber auch Aufzeichnung von Reklamationen haben. Der Entwurf verbessert auch die Verfügbarkeit von klinischen Daten. Der Schutz der Patienten, die an klinischen Studien teilnehmen, wird ebenso verstärkt.
Verschärfte Regeln für Hochrisiko-Medizinprodukte
Bestimmte Hochrisiko-Produkte, wie zum Beispiel Implantate, können einer zusätzlichen Überprüfung durch Experten unterzogen werden, bevor sie auf den Markt gebracht werden dürfen. Expert Panels und Laboratorien werden eine entscheidende Rolle spielen, um Know-How und Beratung zu klinischen Themen an benannte Stellen, zuständige Behörden und Hersteller weiterzugeben.
Die neuen EU-Vorschriften betreffen auch ausdrücklich bestimmte Geräte ohne einen medizinischen Zweck, aber mit ähnlichen Eigenschaften wie medizinische Geräte. Dies betrifft zum Beispiel Füllstoffe und farbige Kontaktlinsen für kosmetische Zwecke.
Mehr Transparenz für die Patienten und eine erhöhte Rückverfolgbarkeit
Es wird eine zentrale Datenbank eingerichtet, um ein verbessertes System für alle relevanten Informationen zu erstellen. Diese Datenbank wird alle relevanten Informationen der beteiligten Unternehmen, der benannten Stellen, der Marktüberwachung, der klinischen Studien und Zertifikate umfassen. Darüber hinaus wird die Datenbank den Patienten, den Fachleuten des Gesundheitswesens und der Öffentlichkeit umfassende Informationen über Produkte in der EU zur Verfügung stellen. Dadurch werden bessere Entscheidungen auf Basis von fundierten Informationen möglich.
Medizinprodukte werden außerdem eine UID-Nummer bekommen, um die lückenlose Rückverfolgbarkeit gewährleisten zu können.
Unser Angebot
Nutzen Sie unsere Informationsangebote:
- Regulatory MedTech-Newsletter
- Auf unserer Website: alle wichtigen Informationen, Links und Dokumente zum Download:
https://www.encotec.at/die-neuen-eu-verordnungen/ - XING Gruppe Regulatory News MEDTECH
Unsere neuen MDR-Veranstaltungsformate:
- Open Evening Lectures in entspannter Kaffeehaus-Atmosphäre: 28.3., 27.4., 30.5. und 20.6.17, jeweils 17:30 bis 20:30 Uhr in Wien. Nähere Infos hier: Open Evening Lectures – die neue MDR / IVDR
- Die neue MDR – Impact Check Workshop (halb- oder ganztags) bei Ihnen vor Ort, um die konkreten Auswirkungen und Maßnahmen zu besprechen.
Ich wünsche Ihnen gutes Gelingen bei der Vorbereitung auf die kommenden Anforderungen und unterstütze Sie gerne dabei!
DI Martin Schmid
Geschäftsführer & Senior-Consultant bei en.co.tec